








































Molecular Genetics
Molecular genetics is the study of molecules and mechanisms involved in genetic inheritance. Archival information molecules are long polymers of deoxyribonucleic acid (DNA) comprising bases in specific sequence. The bases adenine (A), thymine (T), cytosine (C), and guanine (G), function as codon triplets – sequences of three bases that code for specific amino acids or for translation initiation (start codons) or termination (stop codons). Uracil is substituted for thymine in RNA.
The segments of DNA that contain protein-coding instructions are called genes, and these gene sequences comprise a portion of the total genome of a cell. The genome includes both the genes (coding-sequences, domains) and the non-coding sequences – both exons, which include open reading frames, and introns.
Because the 64 possible combinations of GATC code for only the 20 amino acids commonly found in proteins, the code is 'degenerate' (redundant) with more than one triplet combination coding for each amino acid. (This code reduncancy provides hereditary stability by reducing mutation mistakes.) The double helix of DNA comprises paired nucleotide strands with bases hydrogen bonded to complementary bases in the adjacent chain. Adenine pairs with thymine or uracil (A-TU), and cytosine pairs with guanine (CG).
During cellular reproduction, strands of archival DNA are copied or replicated. Transcription is the first step in gene expression – DNA instructions are converted into mRNA codons, rRNAs, miRNAs, and tRNAs. Coding instructions of nucleotide sequences in archival DNA, which have been transcribed and processed into mRNAs are translated into polypeptides and proteins at cytoplasmic ribosomes. Translation is the ultimate step in gene expression, in which archival genetic instructions are converted into specified sequences of amino acids in peptides, polypeptides, and proteins.
In prokaryotic cells – without a nuclear membrane – translation into polypeptides and proteins may begin prior to termination of transcription. The molecular genetics of eukaryotic cells is more complicated than that of prokaryotes. Various molecules of ribonucleic acid (RNA) participate in the transcription of the DNA code into processed mRNA in a series of RNA processing stages including capping, polyadenylation, and pre-mRNA splicing.Following pre-mRNA processing, RNAs undergo extranuclear transfer. Mature RNAs may undergo post-transcriptional modulation (via miRNAs) before translation of the archival DNA instructions into specific sequences of amino acids in the polypeptides and proteins that participate in cellular function and structure. Transfer RNAs (tRNA) deliver specific amino acids to the cytoplasmic ribosomes along the rough endoplasmic reticulum. Ribosomal RNAs participate in assembly of polypeptides and proteins at ribosomes. Here RNAs serve as ribozymes – non-protein enzymes.
A number of processes are involved in control of cellular function through the maintenance of accuracy of genetic inheritance – damage to DNA is repaired, and faulty RNA is destroyed.
DNA damage may result from replication errors, incorporation of mismatched nucleotides (substitution errors – transitions and transversions), oxygen radicals, hydroxyl radicals, ionizing or ultraviolet radiation, toxins, alkylating agents, and chemotherapy agents. A number of vital mechanisms repair DNA damage to bases (including C to T, C to U, and T U mismatch) and to strands, including double strand breaks. All organisms, prokaryotic and eukaryotic, utilize at least three enzymatic excision-repair mechanisms for damaged bases: base excision repair, mismatch repair, and nucleotide excision repair.
Given the importance of mRNA as an information-carrying molecule, faulty pre-mRNAs and mRNAs must be eliminated – they are destroyed by nonsense-mediated decay or nonstop decay:
1. A pre-mRNA made from a mutant gene usually has an exon junction complex (EJC) in the wrong position. This error activates nonsense-mediated decay (NMD) and destroys the pre-mRNA before it can be used to make flawed proteins. There are at least two kinds of NMD: one requires the protein UPF2 and the other does not.
2. Nonstop decay is mRNA turnover mechanism that has none of the properties of normal mRNA turnover or of NMD. A multi-enzyme complex called the exosome is important for nonstop decay. The exosome is the site for binding of a specific adapter protein called Ski7p. Nonstop decay shares none of the enzymes required for nonsense-mediated decay.
Table Mechanisms of Biological Evolution : Gene Regulation in E.coli : | 2 Guide-Glossary

The segments of DNA that contain protein-coding instructions are called genes, and these gene sequences comprise a portion of the total genome of a cell. The genome includes both the genes (coding-sequences, domains) and the non-coding sequences – both exons, which include open reading frames, and introns.
Because the 64 possible combinations of GATC code for only the 20 amino acids commonly found in proteins, the code is 'degenerate' (redundant) with more than one triplet combination coding for each amino acid. (This code reduncancy provides hereditary stability by reducing mutation mistakes.) The double helix of DNA comprises paired nucleotide strands with bases hydrogen bonded to complementary bases in the adjacent chain. Adenine pairs with thymine or uracil (A-TU), and cytosine pairs with guanine (CG).
During cellular reproduction, strands of archival DNA are copied or replicated. Transcription is the first step in gene expression – DNA instructions are converted into mRNA codons, rRNAs, miRNAs, and tRNAs. Coding instructions of nucleotide sequences in archival DNA, which have been transcribed and processed into mRNAs are translated into polypeptides and proteins at cytoplasmic ribosomes. Translation is the ultimate step in gene expression, in which archival genetic instructions are converted into specified sequences of amino acids in peptides, polypeptides, and proteins.
In prokaryotic cells – without a nuclear membrane – translation into polypeptides and proteins may begin prior to termination of transcription. The molecular genetics of eukaryotic cells is more complicated than that of prokaryotes. Various molecules of ribonucleic acid (RNA) participate in the transcription of the DNA code into processed mRNA in a series of RNA processing stages including capping, polyadenylation, and pre-mRNA splicing.Following pre-mRNA processing, RNAs undergo extranuclear transfer. Mature RNAs may undergo post-transcriptional modulation (via miRNAs) before translation of the archival DNA instructions into specific sequences of amino acids in the polypeptides and proteins that participate in cellular function and structure. Transfer RNAs (tRNA) deliver specific amino acids to the cytoplasmic ribosomes along the rough endoplasmic reticulum. Ribosomal RNAs participate in assembly of polypeptides and proteins at ribosomes. Here RNAs serve as ribozymes – non-protein enzymes.
A number of processes are involved in control of cellular function through the maintenance of accuracy of genetic inheritance – damage to DNA is repaired, and faulty RNA is destroyed.
DNA damage may result from replication errors, incorporation of mismatched nucleotides (substitution errors – transitions and transversions), oxygen radicals, hydroxyl radicals, ionizing or ultraviolet radiation, toxins, alkylating agents, and chemotherapy agents. A number of vital mechanisms repair DNA damage to bases (including C to T, C to U, and T U mismatch) and to strands, including double strand breaks. All organisms, prokaryotic and eukaryotic, utilize at least three enzymatic excision-repair mechanisms for damaged bases: base excision repair, mismatch repair, and nucleotide excision repair.
Given the importance of mRNA as an information-carrying molecule, faulty pre-mRNAs and mRNAs must be eliminated – they are destroyed by nonsense-mediated decay or nonstop decay:
1. A pre-mRNA made from a mutant gene usually has an exon junction complex (EJC) in the wrong position. This error activates nonsense-mediated decay (NMD) and destroys the pre-mRNA before it can be used to make flawed proteins. There are at least two kinds of NMD: one requires the protein UPF2 and the other does not.
2. Nonstop decay is mRNA turnover mechanism that has none of the properties of normal mRNA turnover or of NMD. A multi-enzyme complex called the exosome is important for nonstop decay. The exosome is the site for binding of a specific adapter protein called Ski7p. Nonstop decay shares none of the enzymes required for nonsense-mediated decay.
Table Mechanisms of Biological Evolution : Gene Regulation in E.coli : | 2 Guide-Glossary


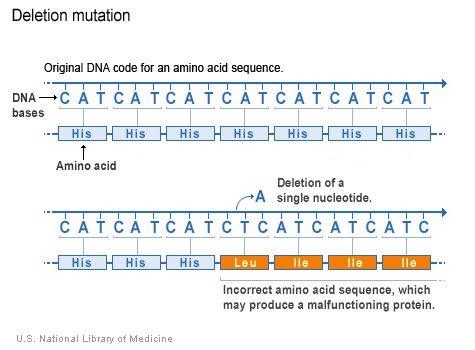
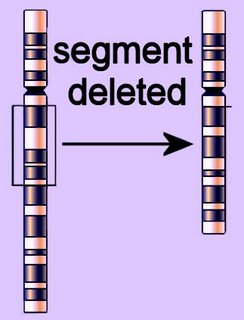 The deletion mutation eliminates one or more nucleotides from a DNA sequence, and may produce a non-functional protein. If the number of deleted bases is not a multiple of 3, then the deletion will cause frameshift, with potentially serious consequences.
The deletion mutation eliminates one or more nucleotides from a DNA sequence, and may produce a non-functional protein. If the number of deleted bases is not a multiple of 3, then the deletion will cause frameshift, with potentially serious consequences.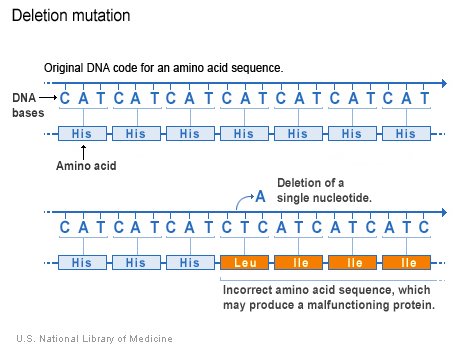


{kind=link}
{kind=link}